Unidade VIII - Biologia Molecular
5. Projetos Genomas
Um dos grandes avanços alcançados nos últimos anos advém do uso da Tecnologia do DNA recombinante, Clonagem, PCR e Sequenciamento do DNA para a obtenção de genomas de organismos como H. influenzae (1ª. bactéria com genoma completo, em 1995), E coli, S. cerevisae, D. melanogaster, C.elegans e outros; e dentre estes, o humano. O objetivo do Projeto Genoma Humano (PGH) foi o de construir um mapa físico detalhado de todo o genoma humano. A HUGO (human genome organization) coordenou os esforços dos geneticistas em todo o mundo para esse fim. O pesquisador James Watson foi o primeiro diretor do projeto. Em junho de 2000, foi anunciado pela HUGO e empresa CELERA (privada), que 96% do genoma havia sido sequenciado e mapeado, e cerca de 25 mil genes foram identificados a partir de 3,1 bilhões de bases.
O potencial uso do conhecimento das sequências levantam questões de cunho ético e social, dependendo de como serão usadas. Por exemplo, existe a possibilidade de identificação de indivíduos com doenças neurogenerativas dominantes associadas com a expansão de nucleotídeos, normalmente CAG (X-frágil, Machado-Joseph, Doença de Huntington). Associada à fertilização in vitro, um casal portador de alelo poderia aceitar ou recusar a implantação de um embrião, após análise de seu DNA e presença do alelo para essas doenças. Será que seria importante que os indivíduos que já sabem que tem pais heterozigotos, poderem escolher se desejam saber se são dominantes e que por volta dos 30 anos poderão apresentar os sintomas? Como preparar psicologicamente as pessoas frente a essas descobertas?
Por outro lado, quando há a descoberta de doenças para as quais já existam medicamentos ou outros métodos de tratamento, ou que indiquem necessidade de mudança de comportamento e alimentação, seria importante o diagnóstico e para que esse seja realizado, é importante o conhecimento das mutações ou polimorfismos que causam o distúrbio. Além disso, o conhecimento da estrutura gênica é importante para o desenvolvimento de diagnóstico e terapias gênicas.
O sequenciamento de curtas sequências RNAs (EST, termo que vem do inglês expressed sequences tags), que foram expressos em uma determinada condição (normal e/ou alterada), é feito após clonagem e montagem de uma biblioteca gênica, ou seja, os cDNAs são utilizados para gerar as ESTs. Como os RNAs mensageiros possuem ORF, os projetos de sequenciamento de ESTs são denominados de ORESTES. Essas sequências de ESTs podem ser completas ou parciais, e, no caso dessas últimas, podem ser alinhadas sobre contigs genômicas.
Sequenciamento de DNA pelo Método de SangerNa década de 1960, as pesquisas do bioquímico Fredrick Sanger permitiram iniciar o sequenciamento do RNA, o que tornou teoricamente possível entender toda a enorme quantidade de informações contidas no DNA, e não apenas exemplos isolados. Isso levou ao real interesse em conhecer a relação entre cada gene e cada característica física, inclusive as doenças de origem genética. O método original de Sanger para o sequenciamento de ácidos nucléicos é um método enzimático, utilizando reações simples e individualizadas de PCR seguida de eletroforese. Por este motivo, e porque nenhuma polimerase inicia o seu trabalho se não tiver disponível uma extremidade 3’OH livre, é necessário para a utilização deste método ter algum conhecimento da sequência da extremidade 5’ da região a sequenciar. Com este conhecimento, é possível desenhar um primer complementar da cadeia a sequenciar, que por hibridização com esta vai fornecer a necessária extremidade 3’-OH livre.
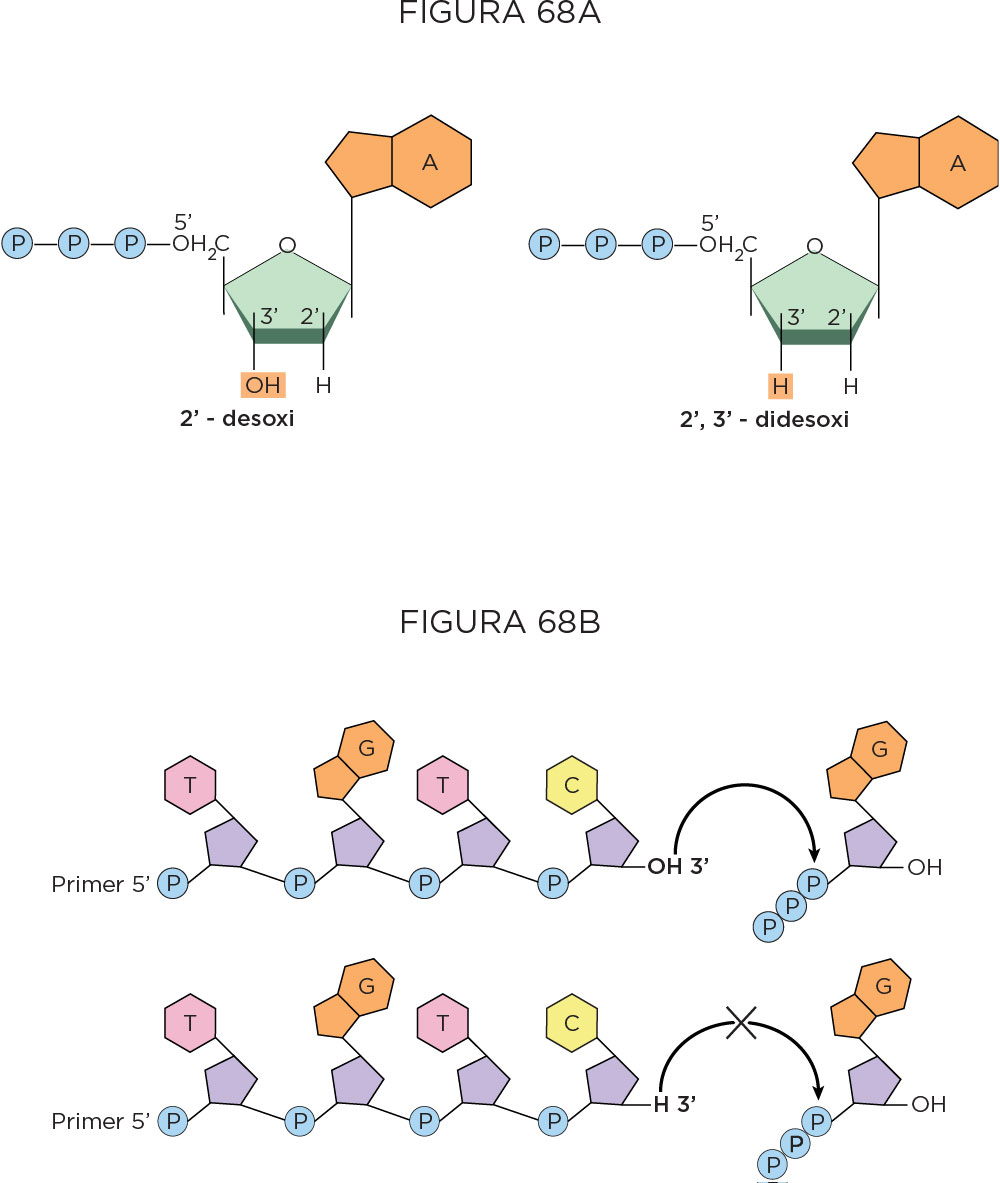
A reação prossegue, incorporando nucleotídeos até que um nucleotídeo terminador (didesoxi) seja incorporado. Os produtos da reação de sequenciamento de Sanger eram separados em géis de poliacrilamida segundo o seu peso molecular. Se em cada reação de sequenciamento for apenas incorporado uma espécie de terminador, cada reação pode servir como revelador da posição em que aparece o respectivo nucleotídeo. A eletroforese em paralelo das quatro reações irmãs de sequenciamento permite então obter um gel clássico, que se lê seguindo os produtos com peso molecular sucessivamente crescente.
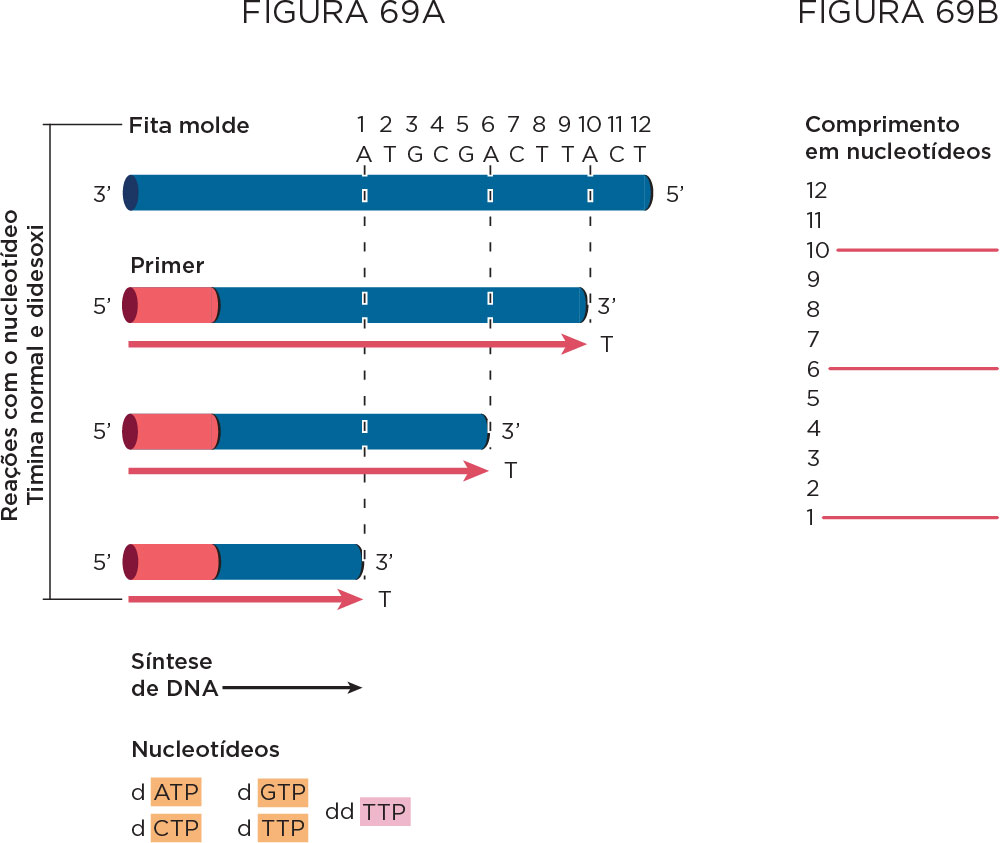
Com o intuito de facilitar a leitura dos géis de sequenciamento, tornando ao mesmo tempo mais precisas as diferenças de migração em gel e rápidas, as quatro reações de sequenciamento foram incorporadas numa só, utilizando, para isso, terminadores modificados. A modificação consiste na marcação de cada espécie de terminador com um fluorocromo (sinal emitido como cores diferentes e captada pelo software), o que permite que cada produto de PCR específico emita luz num comprimento de onda bem determinado e a leitura dos géis é feita por equipamentos específicos capazes de analisar os comprimentos de onda emitidos.
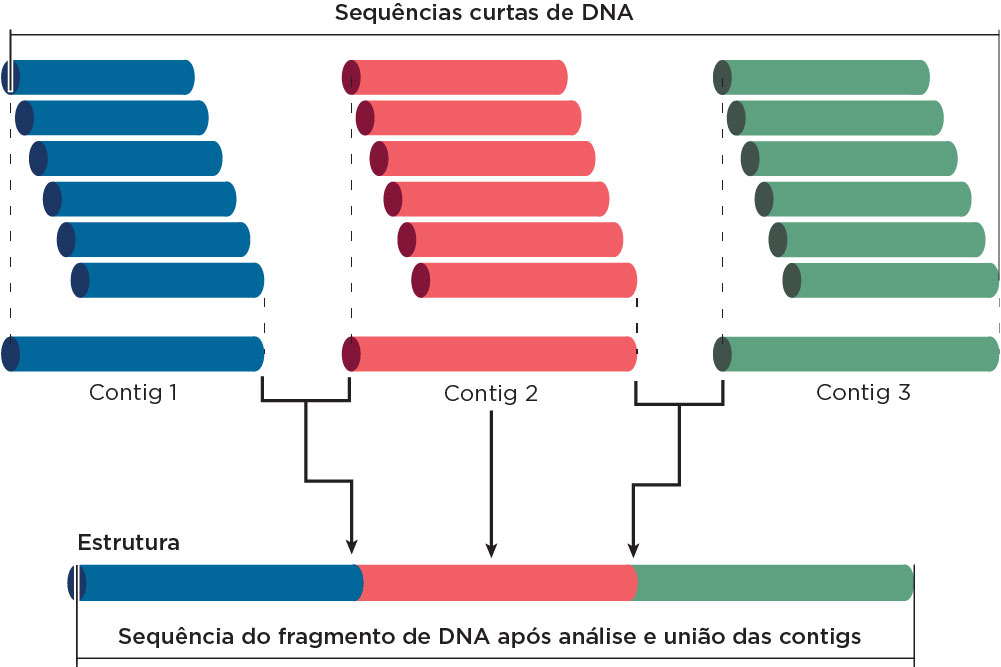
No caso dos sequenciamentos em larga escala, como os que são feitos para obtenção dos genomas, foram sendo desenvolvidos sofisticados programas de computador, que foram desenvolvidos para organizar as sequências de DNA obtidas em sequências contínuas maiores, denominadas contigs. Assim, as leituras contendo sequências idênticas, são consideradas como sobreposições e são reunidas, formando contigs maiores e quanto maior o número de sequências com partes sobrepostas, maior será a sequência obtida e menor a quantidade de lacunas nessa sequência que está sendo organizada. Nos projetos genomas foram feitos alinhamento dos clones, ordenamento e posicionamento nos cromossomos, seguida da caracterização de sequências gênicas.
Bioinformática
Atualmente o termo “Bioinformática” tem sido normalmente utilizado para se referir à análise computacional de dados genômicos, o que inclui também análises de interações DNA-DNA, DNA-RNA, RNA-RNA, proteína-proteína, proteína-ácido nucleico, sendo referida ainda como Biologia Computacional. O sentido desse termo pode ser bem mais amplo, incluindo tudo relacionado à biologia que possa ser auxiliado pela computação.
Porém, é natural relacionar análise de dados genômicos à computação, devido à enorme quantidade de informação, dificultado pelo tratamento manual. As bases atuais de dados genômicos como http://www.ncbi.nlm.nih.gov e http://www.expasy.org, dentre outros, ajudam pesquisadores a entender as funções biológicas, as estruturas químicas e a história evolutiva de organismos. Quando uma nova sequência de bases de nucleotídeos, ou aminoácidos é decifrada, esta pode ser comparada às sequências cadastradas na base, a fim de encontrar sequências relacionadas a ela, o que pode ajudar a revelar algumas propriedades da sequência em questão. Esta área de pesquisa tem se tornado cada vez mais importante devido ao rápido crescimento das bases de dados e sua análise manual pode ser impraticável.
O blast é uma das ferramentas presentes no site do NCBI (National Center for Biotechnology Information), a qual permite, a partir de uma curta sequência de nucleotídeo ou proteína (query sequence), realizar a busca de um ou vários genomas, fornecendo a probabilidade de que aquela seja a sequêcia procurada (hits) de todas as sequências de ácidos nucleicos e proteínas previstas relacionadas a sequência pesquisada.
Variabilidade Genômica, sequências repetitivas e análise de fingerprinting
O DNA em tandem (sequencial) e hipervariável, também conhecidos como DNA satélite, envolve sequências repetidas, uma seguida de outra e agrupadas. As famílias de DNA satélite variam quanto à localização no genoma e comprimento das unidades repetidas que constituem a sequência, formando algumas classes, tais como:
- Minissatélites ou VNTRs (variable number tandem repeats) correspondem às repetições em blocos (tandem) geralmente maiores que sete (CAGT/CAG…) e representam polimorfismos abundantes nas populações. As análises de fingerprintig por Southern-blot analisam os blocos de VNTRs presentes nos indivíduos.
- Microssatélites ou STR (short tandem repeats) são pequenas sequências repetidas denominadas de mononucletotídeos se a mesma base se repete (repetições de poliadeninas), apresentando dinucleotídeos com repetições de duas bases (citosina-adenina/C-A) e até o agrupamento curto de mais bases que estão espalhados pelo genoma, na sua maioria em regiões não codificadoras. As análises de fingerprintig por PCR analisam os blocos de repetições STRs entre os indivíduos, sendo atualmente, amplamente empregada nos exames de DNA, como os de paternidade.
Além do DNA em tandem, há o DNA repetitivo disperso. Nessa classe, estão os elementros de transposição chamados de transposons. Quando há um intermediário de RNA, esses elementos são chamados de retrotransposons Nesses elementos as repetições podem ser encontradas como cópias únicas distribuídas pelo genoma, ou agrupadas, conferindo-lhes a capacidade de “se mover” de um local para outro no genoma, normalmente deixando uma cópia original no lugar, o que faz com que possam se acumular por todo o genoma, causando mutações e doenças genética em humanos. No entanto, o movimento dos transposons é relativamente raro nas células humanas, mas esses elementos foram tão bem-sucedidos na sua propagação que compreendem cerca de 50% do genoma humano. Os transposons parecem ter um papel mais estrutural, por exemplo, DNA intergênico, do que associado à codificação de proteínas (menor que 2% no homem).
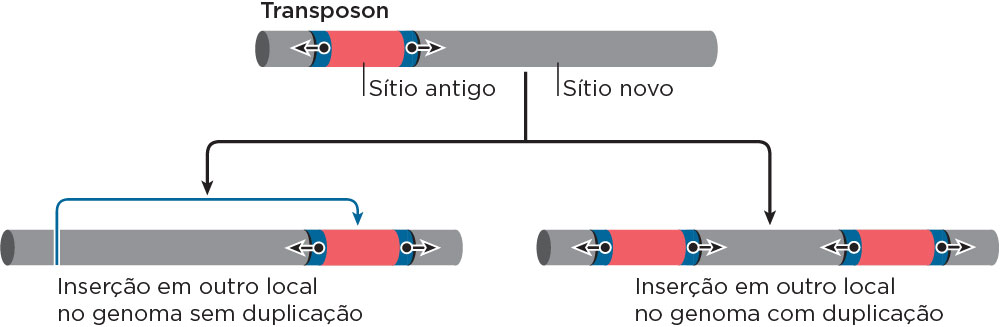
Em plantas parece que esses elementos ocupam uma proporção maior no genoma enquanto em organismos mais simples, como bactérias e fungos, seu número é muito menor. A grande quantidade dessas sequências repetitivas nos organismos mais complexos indica sua importância na manutenção das funções celulares vitais, não podendo ser considerado como DNA lixo.
Os transposons podem se inserir dentro de genes, prejudicando completamente a função dos mesmos, assim como podem se inserir dentro de sequências gênicas reguladoras, onde sua presença pode provocar alterações na expressão desse gene. Foi a partir das perdas de função e alterações na expressão gênica que os transposon foram descobertos. Na década de 40, a pesquisadora Barbara McClintock evidenciou a presença dos transposon em milho por meio de variação na coloração das sementes na espiga. Existem muitos tipos diferentes de elementos de transposição e de uma forma mais didática, esses podem ser divididos em três classes por compartilharem características comuns na estrutura e em sua organização geral, bem como nos mecanismos de transposição, que são os transposon de DNA, retrotransposons semelhantes a vírus e retrotransposons com poli A.
Os transposons de DNA permanecem como DNA do começo ao fim de um ciclo de recombinação, se deslocando utilizando mecanismos que envolvem a clivagem, devido à presença de um gene par a transposase, seguido da religação das fitas de DNA pela presença dos sítios de recombinação (sequências repetidas específicas e invertidas de DNA) que delimitam o gene transposase, além de codificar outras proteínas que participam da recombinação. Os sítios de recombinação presentes nas extremidades terminais invertidas variam em extensão de 25 a algumas centenas de pares de bases, não sendo repetições exatas de sequência e possuem as sequências de reconhecimento da recombinação pelas transposases (algumas vezes chamadas de integrases). Diversos transposons de DNA bacterianos contêm genes que codificam proteínas que conferem resistência a um ou mais antibióticos. A presença do transposon, portanto, torna a célula hospedeira resistente a esse antibiótico.
Os transposons de DNA que contêm o gene da transposase são os chamados de transposons autônomos e aqueles mais simples, que possuem as sequências invertidas, mas não contêm o gene da transposase na sua sequência, são denominados transposons não autônomos. Esses elementos possuem apenas as repetições terminais invertidas, que são as sequências que atuam em cis, necessárias à transposição. Em uma célula que tenha também um transposon autônomo, que codifique uma transposase, esta irá reconhecer também as repetições terminais invertidas do elemento não autônomo, permitindo sua transposição. Entretanto, na ausência desse transposon “auxiliar” (para fornecer a transposase), os elementos não autônomos permanecem estáticos, incapazes de deslocamento.
Os retrotransposons semelhantes aos vírus, também chamados de retrotransposons LTR, constituem elementos móveis que se deslocam para novos locais do DNA utilizando um intermediário temporário de RNA. Os retrotransposons também possuem sequências terminais repetidas e invertidas que são os sítios de ligação e ação da transposase. As repetições terminais invertidas estão inseridas em sequências repetidas mais longas, organizadas nas duas extremidades do elemento, como repetições diretas e são denominadas longas repetições terminais ou LTRs (long terminal repeats). Esses retrotransposons codificam proteínas necessárias para a sua mobilidade como a integrase/transposase e a transcriptase reversa. A transcriptase reversa (RT) é necessária à transposição, pois esta ocorre através de um intermediário de RNA. Como esses elementos convertem RNA em DNA, o reverso do trajeto normal do fluxo da informação biológica (DNA para RNA), eles são conhecidos como “retro” elementos. A distinção entre retrotransposons semelhantes aos vírus e retrovírus é que o genoma de um retrovírus é empacotado em uma partícula viral que sai da sua célula hospedeira e é capaz de infectar uma nova célula. Ao contrário, os retrotransposons podem se deslocar apenas para novos sítios no DNA dentro de uma mesma célula.
Os retrotransposons com poli(A), também descritos como retrotransposons não-LTR, não apresentam as repetições terminais invertidas características presentes nas outras duas classes de transposons, mas as duas extremidades do elemento móvel apresentam sequências distintas, sendo que uma extremidade é chamada de 5’-UTR enquanto a outra extremidade apresenta uma região chamada 3’-UTR, seguida por uma região poli(A). Também apresentam as ORFs que codificam a transposase e a RT. Normalmente, os retrotransposons com poli(A) se apresentam em ambas as formas, autônoma ou LINEs e não autônoma ou SINES.
Os LINES foram identificados primeiramente como uma família de sequências repetidas e o termo vem do inglês como longo elemento nuclear intercalado (Long Interspersed Nuclear Element), são abundantes nos genomas de vertebrados, correspondendo a cerca de 20% do genoma humano, com tamanho médio de 6.000 pb. O elemento L1 é um dos LINEs mais bem conhecidos do genoma humano. Além de promover sua própria mobilidade, os LINEs também fornecem as proteínas necessárias para a transcrição reversa e para a integração de outras classes de sequências repetidas relacionadas, os retrotransposons com poli(A) não autônomos, conhecidos como curtos elementos nucleares intercalados, os SINES (Short Interspersed Nuclear Elements), representado pela sequência Alu, bem difundida no genoma humano e que pode ser clivada pela endonuclease de restrição denominada Alu I.
Como curiosidade, as similaridades dos retrotransposons com retrovírus vão além de sua capacidade de inserir no genoma, como visto nos elementos de transposição de leveduras, os Ty (Transposons in Yeast), em que o RNA de Ty, é encontrado nas células empacotado em uma espécie de partícula viral, no entanto não podem sair e infectar novas células como fazem os vírus. De forma diferente da maioria dos transposons, os Ty se integram, preferencialmente, em regiões cromossômicas específicas, como, por exemplo, os elementos Ty1 de S.cerevisae quase sempre se transpõem no DNA próximos à região promotora de genes transcritos pela RNA polimerase III, que transcreve especificamente os genes de RNAt.
No genoma, existem cópias adicionais de sequências altamente relacionadas a diversos genes celulares, além das cópias repetitivas, as quais parecem ter perdido seus promotores e introns e, frequentemente, contêm interrupções próximas das extremidades 5’. Essas sequências são conhecidas como pseudogenes processados e, normalmente, não são expressos na célula. Esses pseudogenes são, muitas vezes, flanqueados por pequenas repetições no DNA-alvo.
Por fim, existe ainda uma parte do genoma que também não está associada à expressão de proteínas ou a RNAs estruturais, chamadas de DNA intergênico, correspondendo a mais de 60% do genoma humano e não apresenta função conhecida. Assim como nas classes descritas anteriormente, existem os DNAs intergênicos únicos e os repetitivos. Cerca de 25% do DNA intergênico é único, compreendendo regiões aparentemente não funcionais, incluindo genes mutantes não funcionais, fragmentos de genes e pseudogenes. Os genes mutantes e os fragmentos de genes surgem da mutagênese aleatória simples ou erros de recombinação do DNA.